Категории:
ДомЗдоровьеЗоологияИнформатикаИскусствоИскусствоКомпьютерыКулинарияМаркетингМатематикаМедицинаМенеджментОбразованиеПедагогикаПитомцыПрограммированиеПроизводствоПромышленностьПсихологияРазноеРелигияСоциологияСпортСтатистикаТранспортФизикаФилософияФинансыХимияХоббиЭкологияЭкономикаЭлектроника
Способы получения галогеналканов
Способы получения органических галогенидов можно объединить в две группы: реакции замещения и реакции присоединения. Выбор пути и условий реакции зависят от строения углеродного скелета галогенируемой молекулы и того атома или группы атомов, которые подвергаются замещению (уходящие группы). Водород в алканах и алкильных группах, связанных с алкеновым или ареновым фрагментом замещают действием галогена при освещении или нагревании, т.е. в условиях свободнорадикальной реакции.

Галогеном можно заместить не только водород, но также некоторые функциональные группы. Эти превращения протекают по ионному механизму. Галогеналканы часто получают из соответствующих спиртов. Для этого используют различные реагенты нуклеофильного характера. По той причине, что гидроксид-ион – плохая уходящая группа, ее подвижность необходимо повышать путем протонированя (1), комплексообразования (2), или превращения в эфир неорганической кислоты (3).

Поэтому для получения галогеналканов применяют сильнокислую среду либо используют галогенангидриды неорганических кислот: тионилхлорид, хлорокись фосфора, пентахлорид фосфора и др. Иодалканы действием иодоводородной кислоты (аналогично HBr) получить с удовлетворительнми выходами не удается из-за сильной восстановительной способности иодоводорода, который со значительным выходом превращает образующийся иодалкан в алкан.

Более удобно иодалканы получать из хлоралканов по обменной реакции с иодидом натрия (реакция Финкельштейна). Реакцию рекомендуется проводить в ацетоне, т.к. в нем иодид натрия растворим, а образующийся хлорид натрия выпадает в нерастворимый осадок. Это способствует смещению равновесия реакции в сторону образования иодалкана.

Фторалканы получить прямым фторированием не удается, так как активный фтор разрушает молекулу алкана. В качестве одного из путей синтеза фторалканов используют замещение хлора на фтор действием на хлоралкан неорганическими фторидами.

Получение галогенопроизводных посредством реакций присоединения молекул галогенов и галогеноводородов к непредельным соединениям позволяет синтезировать моно-, ди- и полигалогениды.

Реакции нуклеофильного замещения при насыщенном атоме углерода
Реакции нуклеофильного замещения (примеры)
Примеры реакций нуклеофильного замещения весьма многочисленны, при этом в качестве субстрата может выступать не только галогеналкан, но и соединения других классов. В ряду галогеналканов возможны реакции замещения другим галогеном, гидроксилом, алк(ар)оксилом-, нитро-, циано-, аминогруппой и др.

Для замещения галогена цианогруппой применяют цианиды щелочных металлов, реагентом для замещения нитрогруппой служат нитриты щелочных металлов или серебра.
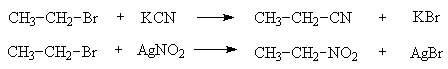
Цианид- и нитрит-ион относятся к амбидентным анионам, т. е. таким, которые имеют по два нуклеофильных центра. Каждый из них образует два изомерных продукта взаимодействия с галогеналканами - нитрил и изонитрил, нитросоединение и алкилнитрит, соответственно.

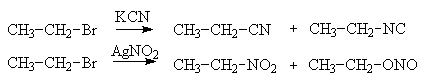
Взаимодействие галогеналкана с аммиаком и аминами приводит сначала к образованию соответствующей аммониевой соли. Последняя превращается в амин при действии основания, роль которого часто выполняет сам амин, взятый в избытке.

Нужно обратить внимание, что данная реакция имеет ограниченное препаративное значение и сопровождается побочным образованием большого количества ди- и триалкиламинов, т.к. алифатические заместители, в большинстве случаев обладающие донорным индуктивным эффектом, повышают нуклеофильность аминогруппы.
Одноатомные спирты
Гидроксипроизводные предельного алифатического ряда называют спиртами (алканолами, например, «этанол, бутанол-2»), т.е названия спиртов образуют путем прибавления окончания -ол к названию соответствующего углеводорода. Кроме официальной номенклатуры используют другие: русскую (по названию углеводородного радикала, например, «этиловый спирт»), тривиальную (например, древесный спирт), карбинольную (карбинол – метанол, а другие спирты – как его производные, т.е. трет-бутанол может быть назван триметилкарбинолом).

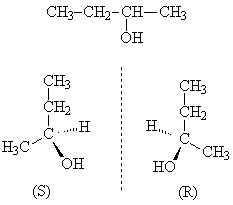
В гомологическом ряду алканолов наблюдается два вида изомерии структурная (изомерия цепи и изомерия положения гидроксила) и оптическая.
Непредельные спирты бывают двух типов: с гидроксилом при двойной связи и аллильные спирты. Ароматические спирты рассматривают как алканолы, в молекулах которых присутствуют ароматические кольца. Те спирты, в которых кратные связи отделены от ОН-группы более чем одним sp3-гибридным атомом углерода, по свойствам не отличаются от обычных алканолов.

| ||
| Виниловый | Аллиловый | Бензиловый |
Нужно отметить, что виниловые спирты, не содержащие сильных акцепторов в молекуле, крайне неустойчивы, они, по сути, являются таутомерной формой соответствующих кетонов, которая присутствует в веществе в незначительной концентрации:

Спиртовая молекула может содержать несколько гидроксильных групп. В соответствии с их количеством различают двух- трех-, многоатомные спирты. Официальные названия многоатомных спиртов образуются путем прибавления окончания -диол, -триол к названию соответствующего углеводорода. Часто применяются тривиальные названия, особенно для 1,2-диолов, называемых гликолями.

Строение и свойства спиртов
Длины связей и валентные углы в молекуле метанола приведены на рисунке

Неподеленные электронные пары атома кислорода расположены на sp3-гибридных орбиталях. ОН-связь имеет такую же длину как в молекуле воды, связь С-Н чуть короче, чем в метане (111,2 пм). Наличие гидроксильной группы придает спиртам свойства как кислот, так и оснований. Кислотность спиртов обусловлена полярностью связи О-Н, о чем позволяют судить величины дипольных моментов, имеющие значения: μС-О = 0,9 D, μ О-Н = 1,5 D. Эти величины указывают на значительное разделение зарядов в гидроксильной группе, благодаря чему водород весьма легко протонизуется. pKaнизших алифатических спиртов близка к pKa воды. В то же время, полярность связи С-О обусловливает возможность замещения гидроксила, о чем будет сказано позднее. Суммарный дипольный момент метанола равен 1,69 D и не соответствует сумме диполей С-О и гидроксила. Это можно объяснить вкладом неподеленных пар электронов кислорода и направлен к кислороду

Неподеленные пары электронов атома кислорода делают спирты слабыми основаниями, способными реагировать с протоном и различными кислотами Льюиса.
Получение спиртов
Способы получения спиртов можно объединить в группы по типам реакций замещения, присоединения и окисления-восстановления. К первой группе относятся гидролиз алкилгалогенидов, сложных эфиров а также реакции замещения других функциональ-ных групп. Гидролиз галогеналканов проводят в условиях реакций нуклеофильного замещения, подбирая их таким образом, чтобы устранить или хотя бы уменьшить возможность протекания побочной реакции элиминирования, приводящей к образо-ванию алкенов.

Широко распространен способ получения спиртов действием азотистой кислотой на первичные алкиламины. В этом превращении первоначально образуется нестабильная алифатическая соль диазония, реагирующая с любым имеющимся в избытке нуклеофилом, в данном случае – с водой.

К реакциям присоединения, приводящим к образованию спиртов, относятся гидратация алкенов, присоединение металлоорганических соединений,к карбонильной группе, гидрирование оксосоединений и карбоновых кислот и др.
Гидратация алкенов протекает в присутствии кислот по правилу Марковникова.

Взаимодействие реактивов Гриньяра и других металлоорганических соединений с альдегидами позволяет получать спирты различного строения и имеет важное препаративное значение. В случае использования формальдегида образуются первичные спирты, из других альдегидов образуются вторичные, а из кетонов – третичные спирты, например:

или

Гидрирование карбонильных соединений проводят как водородом на катализаторе, так и комплексными гидридами металлов (LiAlH4, NaBH4).

Другой способ получения спиртов восстановлением альдегидов и кетонов основан на реакции гидридного обмена, где источником гидрид-иона является алкоголят вторичного спирта, чаще всего изопропилат алюминия (Реакция Меервейна-Пондорфа-Верлея). Эта реакция обратима, поэтому для ее полного протекания необходимо использование большого количества спирта-восстановителя, причем последний должен превращаться в более легко кипящий, чем восстанавливаемое оксосоединение, продукт, чтобы его можно было удалять путем отгонки.

Кроме альдегидов и кетонов, восстановлению до спиртов поддаются также производные карбоновых кислот – хлорангидриды и сложные эфиры.
Можно привести схему восстановления хлорангидрида LiAl(OBut)3H

Более активный алюмогидрид лития восстанавливает в эфирах оксокислот как алкилкарбоксигруппу, так и карбонильный фрагмент, боргидрид натрия сложноэфирную группу не затрагивает.

Свойства спиртов
Кислотность
Величины рКа алканолов, измеренные в водных растворах растут от метанола с увеличением длины углеродной цепи и со степенью замещенности a -углеродного атома.
| Спирт | CH3OH | C2H5OH | (CH3)2CHOH | (CH3)3COH | (CF3)3COH | H2O |
| рКа | 15,5 | 15,9 | 17,1 | 18,0 | 5,4 | 15,64 |
Протон от гидроксильной группы может быть удален действием достаточно сильного основания. Спирты взаимодействуют, например, с металлическим натрием, гидридом натрия, реактивами Гриньяра, образуя соответствующие солеобразные соединения, называемые в общем случае алкоголятами

Как видно из приведенной выше таблицы, константы кислотности низших алканолов и воды близки, поэтому при взаимодействии их со щелочами устанавливается динамическое равновесие, которое смещено влево

Алкоголяты
Алкоголят-ионы являются намного более сильными нуклеофилами чем соответствую-щие спирты, поэтому они способны к взаимодействию с такими слабыми электрофила-ми, с которыми сами спирты не реагируют. Благодаря этому свойству они применяются для получения простых эфиров: при их взаимодействии с алкилгалогенидами образу-ются простые и составные диалкиловые эфиры (синтез Вильямсона, 1852 г.).

Алкоголяты способны к взаимодействию и с другими электрофилами, например, галогенопризводными ароматическими соединениями.

По отношению к спирту это превращение рассматривают как реакцию алкилирования (арилирирования), по отношению к галогенопроизводному как реакцию нуклеофильного замещения.
Основность спиртов
Другим общим свойством спиртов является основность, которая обусловлена неподеленными электронными парами атома кислорода. С кислотами Льюиса спирты образуют оксониевые катионы (в случае протона - гидроксониевые).

Водородные связи
Наличие у спиртового гидроксила одновременно кислотных и основных свойств придает спиртам способность образовывать ассоциаты за счет межмолекулярных водородных связей. Это является причиной более высоких температур кипения спиртов в сравнении с алканами с такой же или даже большей молярной массой. Например, метанол (М=32) кипит при +64оС, бутан (М=58) - при +4оС. Энергия водородной связи невелика (около 20 кДж/моль), но большое количество водородных связей требует значительного расхода энергии на их разрыв при нагревании спирта до температуры кипения. Низшие спирты благодаря водородным связям неограниченно смешиваются с водой.

Нуклеофильность
Электронные пары атома кислорода обусловливают также нуклеофильность спиртов. Это проявляется во взаимодействии с сильными электрофилами. Например, в галогенангидридах и ангидридах кислот действием спиртов замещается и уходит в форме аниона группа при ацильном атоме углерода. Эту реакцию обычно называют ацилированием спиртов, потому что в молекулу спирта вместо водорода вводится ацильная группа .

Реакция ацилирования спиртов карбоновыми кислотами эффективно катализируется сильными минеральными кислотами, Протоны, координируясь с электронными парами карбонильного кислорода карбоксильной группы, повышают электрофильность ацильного углерода. Благодаря тому, что в результате этого происходит превращение кислоты в эфир реакция получила название этерификации.

Образование сложных эфиров при действии ангидридов, галогенангидридов, сложных эфиров происходит легче, когда реакцию проводят в присутствии основания, роль которого состоит в переводе гидроксипроизводного в форму более нуклеофильного алкоголята. Это явление известно как основной катализ ацилирования. В молекулах спиртов можно осуществить замещение гидроксильной группы. Считают, что реакция, по причине полярности связи С-О, идет как нуклеофильное замещение, механизм которого определяется, главным образом, строением спирта. Как и замещение галогена, первичные спирты предпочтительно реагируют по бимолекулярному механизму, третичные - по диссоциативному. Однако в случае спиртов (по сравнению с например, галогеналканами) необходимо увеличить поляризацию С-О-связи, т.к. положительный заряд на a -углеродном атоме спиртов значительно ниже, чем галогенопроизводных. Этого достигают, как правило, проводя реакцию в присутствии кислот (протонных или Льюиса), а также превращая спирты в более активные эфиры неорганических кислот (см. тему "Получение галогеналканов").

Существует и другая причина необходимости проведения реакции замещения гидроксила галогенид-ионами в кислотной среде, которая, как считают, заключается «качестве» уходящей группы. Хорошей уходящей группой считается такая, которая ходе замещения превращается в термодинамически устойчивую частицу. Так, спирт не реагирует с бромидом калия, потому что уходящая группа гидроксид-ион богат энергией и, следовательно, малоустойчив. Напротив, реакция идет в присутствии серной кислоты, так как в результате протонирования гидроксил превращается в гидроксоний и уходит в форме молекулы воды, которая термодинамически намного устойчивее гилроксид-иона. Соляная кислота редко используется для превращения спиртов в хлоропроизводные. Она легко реагирует лишь с третичными спиртами, что объясняется, вероятно, низкой нуклеофильностью хлорид-иона, который способен успешно атаковать "чистый" карбкатион (SN1), но не гидроксониевую частицу, образующуюся в кислой среде из первичного спирта (SN2). Вместе с тем, соляная кислота слабее бромоводородной и поэтому концентрация протонированных молекул спирта и карбокатионов в реакционной среде ниже. Успешному протеканию реакции с соляной кислотой способствует присутствие кислоты Льюиса хлорида цинка, который координируется с электронами кислорода и индуцирует положительный заряд.

Превращение спиртов в эфиры неорганических кислот также повышает их электрофильные свойства. Так, образование диэтилового эфира при нагревании этанола с 96%-ной серной кислотой (130оС) объясняется тем, что в ходе реакции промежуточно возникает этилсерный эфир, который взаимодействует с другой молекулой спирта. По этому способу получения диэтиловый эфир получил название «серный».

Диалкиловые эфиры серной кислоты, называемые диалкилсульфатами, производятся химическими фирмами и широко применяются для алкилирования спиртов, фенолов в присутствии оснований а также аммиака и аминов.
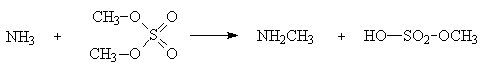
Иодоводородная кислота обладает достаточной кислотностью и иодид-ион является сильным нуклеофилом, но его сильные восстановительные свойства не всегда позволяют с высокими выходами превращать спирты в алкилиодиды.
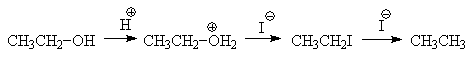
Поэтому замещение гидроксила проводят действием трииодида фосфора, который генерируют из красного фосфора и иода непосредственно в реакционном растворе.

Считают, что на первой стадии реакции трииодид фосфора образует со спиртом эфир (сродство фосфора к кислороду очень велико), который в качестве электрофила участ-вует в реакции нуклеофильного замещения.

Для превращения спиртов в галогеналканы используют также +пентахлорид фосфора и тионилхлорид. Раньше считали, что на первой стадии взаимодействия спирта, например, с тионилхлоридом образуется сложный эфир и затем происходит внутри-молекулярное нуклеофильное замещение (механизм SNi).

При этом механизме должна полностью сохраняться конфигурации при хиральном центре. Однако при термолизе одного из энантиомеров 2-октилхлорсульфита полученный продукт содержит 78% 2-хлороктана с обращенной конфигурацией. Сейчас доказано, что процесс протекает с промежуточным образованием ионных пар.
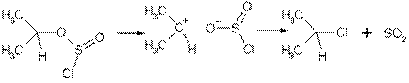
Механизм можно представить следующим образом

Спирты в обычных условиях устойчивы к действию окислителей. Кислородом воздуха на холоду они не окисляются. Тем не менее, сильные окислители превращают их в альдегиды, кетоны и карбоновые кислоты. Первичные спирты окисляются перманганатом калия или бихроматом натрия в сернокислом растворе. Легче всего происходит окисление до карбоновых кислот. Поэтому для получения альдегида, который кипит при более низкой температуре, чем соответствующий спирт, его отгоняют по мере образования для предохранения от дальнейшего окисления в карбоновую кислоту. Другими методами получения альдегидов из спиртов являются использование мягких окислителей (MnO2, SeO2) и превращение альдегида insitu в более устойчивое производное, например, диацетат.

В результате окисления вторичных спиртов при окислении с хорошим выходом образуются кетоны, третичные же спирты окисляются трудно, при этом происходит разрыв углеродной цепи и образуются смеси продуктов.

Спирты превращаются в альдегиды и кетоны также в результате дегидрирования при нагревании с катализаторами Cu, Pt, Pd.

Спирты на холоду или при умеренном нагревании с концентрированными минеральными кислотами образуют сложные эфиры. В большей степени это относится к первичным спиртам; вторичные и особенно третичные при нагревании с кислотами претерпевают дегидратацию, превращаясь в непредельные соединения. Сложные эфиры первичных спиртов с серной или фосфорной кислотой так же образуют алкены при высокой температуре. Так, реакция этанола с серной кислотой при 160 оС приводит к этилену, при 120-130° - к диэтиловому эфиру, а при 70-80° - к моно- или диэтилсульфату в зависимости от соотношения реагентов.

Превращение спирта в этилен называют реакцией бимолекулярного (синхронного) элиминирования (Е2-механизм).

Третичные спирты не образуют эфиров, но очень легко теряют молекулу воды, что происходит, вероятно, по мономолекулярному механизму отщепления (Е1) через промежуточное образование карбкатиона. Например, третичный бутиловый спирт дегидратируется при нагревании с 15%-ной серной кислотой.

Дегидратация вторичных и третичных спиртов может происходить при нагревании с другими кислотами (кислотами Льюиса), а также на поверхности оксида алюминия при высокой температуре. Ориентация отщепления подчиняется правилу Зайцева (образуются наиболее устойчивые, т.е. более замещенные алкены). Как видно из приведенных примеров, реакции элиминирования всегда конкурируют с реакциями замещения. Как и в случае реакций галогеналканов, промежуточное образование карбкатионов не может не привести в ряде случаев к их перегруппировке. Ранее мы встречались с перегруппировкой карбкатионов в более стабильные путем миграции атома водорода к соседнему "ониевому" атому углерода. Однако, мигрировать может не только водород, но так же алкильная или арильная группа. Образующиеся в результате такого превращения алкены отличаются по строению от ожидаемых. Например, довольно известна ретропинаколиновая перегруппировка.
Многоатомные спирты
Первый представитель ряда многоатомных спиртов имеет два атома углерода в молекуле, потому что по правилу, открытому Эльтековым и Эрленмейером, a-дигидрокси-производные в большинстве случаев неустойчивы и, отщепляя воду, превращаются в оксосоединения.

Первый член ряда многоатомных спиртов – этандиол-1,2 обычно называют по тривиальной номенклатуре – этиленгликоль, поэтому все 1,2-двухатомные спирты объединяют под названием гликоли. Трехатомные спирты называются глицеринами по названию самого простого из них - пропантриола-1,2,3. По своим свойствам этиленгликоль, глицерин и другие полиатомные спирты большей частью похожи на алканолы. Однако у них имеются и некоторые специфические свойства, на которые целесообразно обратить внимание. Образование ассоциатов за счет водородных связей проявляется у них в большей степени, чем у алканолов, что связано с бóльшим количеством полярных групп по отношению к длине углеродного скелета. Это является причиной более высоких температур кипения, например, температура кипения этиленгликоля 197 оС, а этанола - 78°С. Этиленгликоль и глицерин представляют собой вязкие жидкости они неограниченно смешиваются с водой, что так же обусловлено высокой степенью ассоциированности. Этиленгликоль является более сильной кислотой (рКа 14,2), чем этанол (рКа 15,9), потому что каждая из гидроксильных групп испытывает -I-эффект соседнего гидроксила . При действии на гликоли сильных оснований образуется два ряда алкоголятов: по одному и по обоим гидроксилам в зависимости от количества основания.

Присутствие двух гидроксильных групп у соседних атомов углерода в молекулах многоатомных спиртов создает благоприятные условия для образования с катионами металлов устойчивых координационных соединений, называемых хелатными комплексами. Образование комплекса меди (II) с глицерином и его строение можно описать схемой

Алкандиоляты щелочных металлов являются сильными нуклеофилами и могут быть легко проалкилированы. Образующиеся при этом простые эфиры находят применение в органическом синтезе в основном в качестве апротонных растворителей (см. тему «Простые эфиры»).

При действии ацилирующих реагентов гликоли образуют два ряда сложных эфиров, глицерины - три. Как и алканолы, многоатомные спирты образуют эфиры не только с карбоновыми, но и с неорганическими кислотами. Например, глицерин встречается в природе в виде сложных эфиров высших алифатических карбоновых кислот, называемых жирами. Растительные жиры состоят преимущественно из глицеридов непредельных олеиновой и пальмитиновой кислот, в животных жирах преобладает ацил насыщенной стеариновой кислоты. Среди эфиров глицерина с минеральными кислотами известен его тринитрат (тринитроглицерин), применяемый как бризантное взрывчатое вещество, его используют также в медицине в качестве сосудорасши-ряющего препарата.

Окисление этиленгликоля, в зависимости от природы окислителя, дает различные продукты.

Действие йодной кислоты на 1,2-диолы приводит к разрушению углеродного скелета и образованию карбонильных соединений (реакция периодатного расщепления). Идентификация образующихся при этом альдегидов используется для установления структуры в химии углеводов.

Этиленгликоль относительно легко дегидратируется. Нагревание этиленгликоля с серной кислотой дает в результате межмолекулярной дегидратации 1,4-диоксан, тогда как в присутствии хлорида цинка реакция протекает по внутримолекулярному механизму, при этом образуется ацетальдегид.
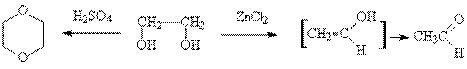
Образование ацетальдегида объясняют тем, что комплекс хлорида цинка с одним из гидроксилов распадется, давая карбокатион, который в результате отщепления протона превращается в виниловый спирт. Последний в кислой среде быстро перегруппи-ровывается в более устойчивую таутомерную форму – ацетальдегид (см. тему «Карбонильные соединения»). В отсутствие же кислот виниловый спирт все-таки удается получить, однако период его "полуизомериции" составляет не больше 30 мин.

Дегидратация глицерина действием гидросульфата калия при нагревании приводит к образованию непредельного альдегида – акролеина. Это превращение имеет механизм, аналогичный описанному выше.

Дегидратация дитретичных и дивторичных гликолей, катализируемая серной кислотой, кислотами Льюиса (BF3 и др.) включает в себя стадию миграции алкильной, арильной группы или гидрид-иона. В результате этой перегруппировки образуются карбониль-ные соединения (пинаколиновая перегруппировка).

Гидроксильные группы многоатомных спиртов, как и в алканолах, могут быть замещены на галоген. Так, этиленгликоль при действии соляной кислоты превращается в этиленхлоргидрин, который переходит в дихлорэтан при обработке пентахлоридом фосфора.

Фенолы
Фенолы содержат гидроксил непосредственно связанный с атомом углерода ароматического кольца. По числу гидроксильных групп, присоединенных к кольцу, фенолы подразделяются на одно-, двух- и многоатомные.
Изомерия фенолов обусловлена взаимным положением заместителей в бензольном кольце.
орто-крезол  мета-крезол
мета-крезол  пара-крезол
пара-крезол 
Получение
Фенол в промышленности получают:
1) окислением изопропилбензола (кумола) в гидроперекись с последующим разложением её серной кислотой.
C6H5(C3H7-i) + O2 ® C6H5(i-C3H6(O-OH) ;
C6H5(i-C3H6(O-OH) + H2SO4 ® C6H5OH + (H3C)2C(O)
2) из бензола по способу Рашига:
C6H6 + HCl + O2 ®  + H2O
+ H2O
+ H2O(Cu2+) ®  + HCl
+ HCl
Химические свойства
1. Кислотные свойства у фенолов выражены сильнее, чем у спиртов. В результате сопряжения неподелённой пары электронов кислородного атома с p- электронной системой ароматического кольца, электронная плотность О-атома перемещается частично на связь С–О, увеличивая при этом электронную плотность в бензольном ядре (в особенности в орто- и пара-положениях). Электронная пара связи О–Н сильнее притягивается к атому кислорода, способствуя тем самым созданию большего положительного заряда на атоме водорода гидроксильной группы и, следовательно, отщеплению этого водорода в виде протона.
 Hd+
Hd+ 
Фенолы, в отличие от спиртов, образуют феноляты не только при взаимодействии с активными металлами, но и с водными растворами щелочей.
C6H5OH+2Na ® 2C6H5ONa(фенолят натрия) + H2
C6H5OH+NaOH ® 2C6H5ONa + H2O
Образование фиолетового окрашивания при добавлении раствора FeCl3 служит качественной реакцией, идентифицирующей фенолы:
3C6H5OH+ FeCl3 «(C6H5OH)3Fe + 3 HCl
Фенолы – очень слабые кислоты, поэтому феноляты легко гидролизуются и разрушаются не только сильными, но и очень слабыми кислотами:
C6H5O Na+ H2SO4 ® C6H5OH+ NaHSO4
C6H5O Na + CO2 + H2O ® C6H5OH + NaHCO3
Фенолы образуют простые эфиры (синтез Вильямсона) и сложные эфиры
C6H5O Na + Br-CH2–CH3 ® C6H5OCH2–CH3 + NaBr
и сложные эфиры: C6H5OH + Cl—(О)C–CH3 ® C6H5O(О)C–CH3 + HCl
(хлорангидрид укс.кислоты) (фениловый эфир укс.кислоты)
2. Реакции бензольного кольца. Гидроксильная группа у фенолов является очень сильным орто- и пара-ориентантом. Реакции замещения атомов водорода в бензольном кольце протекают в более мягких условиях, чем у бензола. При бромировании и нитровании могут быть получены 2,4,6-тризамещенные производные
OH OH OH
 3HNO3 H2SO4
3HNO3 H2SO4  + 3Br2 –––®
+ 3Br2 –––®  + 3HBr
+ 3HBr
NO2 Br
2,4,6–тринитро-фенол (пикриновая кислота) 2,4,6–трибромфенол
3. Гидрирование.
OH OH
 + 3H2(Ni) ––®
+ 3H2(Ni) ––®  (циклогексанол)
(циклогексанол)
Применение
Фенол применяется при производстве фенолформальдегидных смол, в фармацевтической промышленности и как антисептик (карболовая кислота).
Гидрохинон (1,4-диоксибензол) – проявитель в фотографии.
Простые эфиры
К простым эфирам относятся соединения, в которых два углеводородных радикала связаны атомом кислорода. По рациональной номенклатуре они называются по названию соответствующих радикалов: диэтиловый эфир, метилэтиловый эфир, дифениловый эфир. Первый иногда называют серным эфиром (по способу получения) или просто эфиром. Систематическая номенклатура рассматривает простые эфиры как производные алканов ("метоксиэтан" вместо "метилэтиловый эфир"). Иногда применяется "оксидная" номенклатура: ("дифенилоксид" вместо "дифениловый эфир"). Распространены тривиальные названия: анизол (метилфениловый эфир), фенетол (этилфениловый эфир).

Для циклических эфиров номенклатурой IUPAC зарегистрированы тривиальные названия, например:
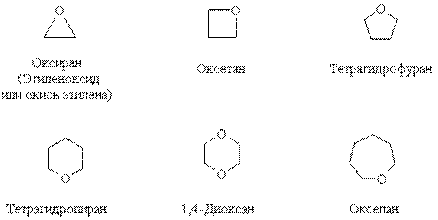
Получение
Простые эфиры образуются из спиртов при действии кислот, обладающих водоотнимающими свойствами, таких как H2SO4 или полифосфорная кислота.
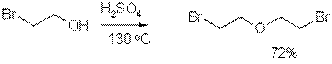
Реакция Вильямсона открывает более широкие возможности, т.к. позволяет синтезировать несимметричные эфиры.

Эта реакция стала интенсивно использоваться для синтеза открытых в 60-е годы краун-эфиров.

Свойства
Химические свойства простых эфиров обусловлены присутствием атома кислорода, связанного с двумя С-атомами. Две несвязывающих пары электронов кислорода обусловливают основность (по Льюису) простых эфиров. Низшие представители сравнительно неплохо растворимы в воде (водородные связи), с сильными кислотами они образуют гидроксониевые соли. Поэтому эфир растворим в концентрированной соляной кислоте и выделяется при ее разбавлении.

Кислоты Льюиса с простыми эфирами дают солеобразные оксониевые соединения, которые называют эфиратами. Кислота и основание связаны донорно-акцепторной связью.

Комплексы эфиров с трифторидом бора особенно стабильны: Сам BF3 при комнатной температуре – газ, диэтиловый эфир кипит при 34.5°С, тогда как их комплекс перегоняется без разложения при 129°С. Несмотря на полярность связи С-О в молекулах простых эфиров (m=1,6D), они не реагируют с нуклеофилами. Очевидно, это обусловлено тем, что в ходе расщепления должно было бы произойти вытеснение более сильного основания - алкоголят-иона (плохая уходящая группа). Напротив, в сильнокислой среде, где плохая уходящая группа алкоксид-ион превращается в хорошую уходящую группу (молекулу спирта), простые эфиры расщепляются действием сильных нуклеофилов. Так, эфиры расщепляются до соответствующего спирта и алкилгалогенида под действием HBr или HI (но не HCl):



Простые эфиры легко вступают реакции свободно-радикального замещения по a-углеродному атому. Так, получение хлордиэтилового эфира ведут действием хлора на эфир при -20 оС.

Это объясняют тем, что в результате отрыва атома водорода возникает частица, стабилизированная за счет нахождения неспаренного электрона в поле атома кислорода ("три электрона в поле двух атомов").

Это же обстоятельство является причиной легкости окисления простых эфиров кислородом по свободно-радикальному механизму. В следствие этого пары эфиров образуют с воздухом взрывчатую смесь. Во время хранения эфира в нем накапливаются гидропероксиды, которые, благодаря более высокой температуре кипения, остаются в перегонной колбе и в конце перегонки могут взрываться. Поэтому эфиры перед перегонкой обрабатывают раствором солей двухвалентного желе
Последнее изменение этой страницы: 2016-07-23
lectmania.ru. Все права принадлежат авторам данных материалов. В случае нарушения авторского права напишите нам сюда...