Категории:
ДомЗдоровьеЗоологияИнформатикаИскусствоИскусствоКомпьютерыКулинарияМаркетингМатематикаМедицинаМенеджментОбразованиеПедагогикаПитомцыПрограммированиеПроизводствоПромышленностьПсихологияРазноеРелигияСоциологияСпортСтатистикаТранспортФизикаФилософияФинансыХимияХоббиЭкологияЭкономикаЭлектроника
Гетерогенный катализ: основные стадии, энергетический профиль.
Катализ– изменение скорости химической реакции в присутствии катализаторов. Катализатор– вещество, участвующее в реакции и изменяющее ее скорость, но остающееся неизменным после того, как химическая реакция заканчивается. Ингибитор – катализатор, замедляющий реакцию. Гомогенные реакции– реагенты и катализатор находятся в одной фазе. Гетерогенные – реагенты и катализатор находятся в разных фазах. Реакция происходит на поверхности катализатора.
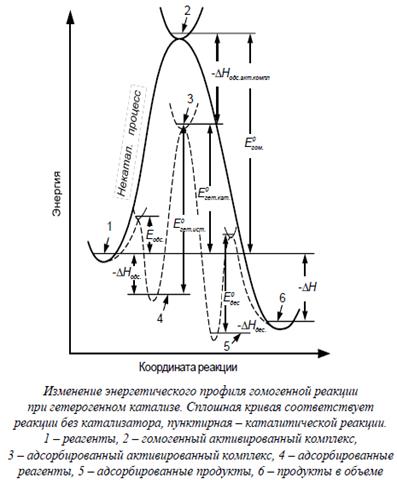
В каталитических реакциях, протекающих на поверхности твердого катализатора, можно выделить пять основных стадий:
1) диффузия вещества к поверхности катализатора;
2) обратимая адсорбция (повышение концентрации одного вещества (газ, жид) у пов-ти другого вещества (жид, тв.тело) вещества;
3) реакция на поверхности, в адсорбционном слое;
4) обратимая десорбция (удаление адсорбированного вещества с поверхности адсорбента) продуктов с поверхности;
5) диффузия продуктов реакции от поверхности в объем.
Общая скорость каталитической реакции определяется скоростью самой медленной из этих стадий.
Если не рассматривать диффузию и считать, что равновесие адсорбция  десорбция устанавливается быстро, то скорость каталитической реакции определяется скоростью реакции в адсорбционном слое, где роль реагента играют свободные адсорбционные центры. Простейший механизм гетерогенного катализа описывается схемой:
десорбция устанавливается быстро, то скорость каталитической реакции определяется скоростью реакции в адсорбционном слое, где роль реагента играют свободные адсорбционные центры. Простейший механизм гетерогенного катализа описывается схемой:
S(газ)+адс.центр k дес↔k адс S(адс)→k1 P(адс)→k2 Р(газ)
На энергетическом профиле реакции, протекающей на поверхности катализатора, появляются дополнительные максимумы и минимумы, связанные с процессами адсорбции реагентов (1 4), десорбции продуктов (5 6) и химической реакции в адсорбционном слое (4 5).
4), десорбции продуктов (5 6) и химической реакции в адсорбционном слое (4 5).
В общем случае уравнение химической реакции на поверхности можно записать следующим образом:

Согласно закону действующих масс для гетерогенной реакции, скорость реакции пропорциональна произведению степеней заполнения поверхности реагентами и свободными центрами:
 , где степени заполнения θ при условии адсорбционного равновесия определяются по уравнению Ленгмюра:
, где степени заполнения θ при условии адсорбционного равновесия определяются по уравнению Ленгмюра:

(KL – константы адсорбционного равновесия, p – парциальные давления). Таким образом, скорость гетерогенной каталитической реакции определяется давлениями не только реагентов, но и продуктов реакции.
Расчёт электродных потенциалов по уравнению Нернста.
E = E° + RT/nF * ln(aox/ared)
n – число электронов,
aox – активность окисленной формы,
ared – … восстановленной. Для любого чист.в-ва активность равна 1.
F=96500 Кл/моль – постоянная Фарадея.
Билет 2.
Электронное состояние атома как целого. Квантовые числа. Атомные
Термы.
2. Энтропия и 2-й закон термодинамики. Термодинамическое и статистическое определения энтропии, их взаимосвязь.
Второй закон термодинамики Существует экстенсивная функция состояния термодинамической системы – энтропия (S). При протекании в изолированной системе обратимых процессов эта функция остается неизменной, а при необратимых – увеличивается: dSU,V≥0.
Из этого следует, что после завершения релаксационных процессов при состоянии термодинамического равновесия энтропия изолированной системы достигает своего максимума: dSU,V=0, d2S<0.
Понятие энтропии было введено в термодинамику Р. Клаузиусом. Неравенство Клаузиуса связывает изменение энтропии с количеством теплоты δQ, которым система обменивается с окружением при температуре Т:
 .
.
Источником необратимого процесса может быть диффузия, расширение системы при существовании разности давлений между ней и окружающей средой, теплопередача при разных температурах, самопроизвольные химические реакции в объеме системы и другие диссипативные процессы, связанные с необратимым превращением работы в теплоту.
Энтропия – внутреннее свойство термодинамической системы; согласно постулатам термодинамики, при равновесии она является функцией внутренней энергии и внешних переменных.
Абсолютное значение энтропии, полученное при интегрировании неравенства Клаузиуса для обратимых процессов, известно с точностью до постоянной интегрирования (S0):

Значение этой постоянной устанавливается третьим законом термодинамики: при нулевой абсолютной температуре энтропия любых веществ, находящихся в равновесном состоянии, имеет одно и то же значение, не зависящее от фазового состояния вещества. В изотермических процессах, происходящих при T=0, энтропия не зависит ни от обобщенных сил, ни от обобщенных координат.
Так как при 0 К энтропия всех веществ одинакова, то конкретное значение S0 несущественно и его можно принять равным нулю (постулат Планка): при абсолютном нуле все идеальные кристаллы имеют одинаковую энтропию, равную нулю. Постулат Планка позволяет ввести понятие абсолютной энтропиивещества, т.е. энтропии, отсчитанной от нулевого значения при Т=0.
Статистическое определение энтропии основано на идее о том, что необратимые процессы в термодинамике вызваны переходом системы в более вероятное состояние, поэтому энтропию можно связать с вероятностью:
S=k ln W,
где k – постоянная Больцмана (k=R/NA), W – так называемая термодинамическая вероятность, т.е. число микросостояний, которые соответствуют данному макросостоянию системы. Формулу называют формулой Больцмана. С увеличением количества молекул и числа доступных уровней энергии термодинамическая вероятность резко увеличивается, так что для обычных молекулярных систем при повышении температуры и разупорядоченности энтропия возрастает. Верно и обратное.
Вероятностное опр. S= – k Σi pilnpi
Фотохимические реакции. Законы фотохимии. Квантовый выход. Примеры фотохимических реакций.
Энергия одного кванта излучения связана с длиной волны соотношением:

где h – постоянная Планка.
Согласно первому закону фотохимии (Гротгус (1817), Дрепер (1830)), фотохимическое превращение может происходить только под действием того света, который поглощается веществом.
Второй закон фотохимии (Штарк и Эйнштейн (1912)): каждый поглощенный фотон вызывает фотохимическое возбуждение одной молекулы. Этот закон нарушается в сильных световых полях, где происходят многоквантовые процессы и одна молекула может поглотить несколько квантов излучения.
При поглощении видимого или УФ света молекула переходит в электронно возбужденное состояние: M+hν→M*.
Поглощение света может привести к разнообразным химическим превращениям электронно-возбужденной молекулы. Примеры первичных фотохимических реакций:
M+hν→M* → (фотохимич.р-я)
→ продукты (1);
→ M**→M+hν1 (фосфоресценция);
→ M+hν (флоуресценция);
→ тушение.
Эффективность фотохимической реакции характеризуют квантовым выходом, который равен отношению числа прореагировавших молекул к числу поглощенных фотонов:

Все фотохимические реакции по значению квантового выхода можно разбить на три группы.
1. φ =1, например образование бромциклогексана или перекиси водорода (1).
2. φ <1, например разложение ацетона или аммиака. Такое значение квантового выхода свидетельствует о том, что в результате первичного процесса образуются устойчивые молекулы, и фотохимическая реакция на этом заканчивается.
3. φ>>1. Если же первичная реакция приводит к появлению реакционно-способных частиц, например, свободных радикалов, то возможны вторичные процессы – цепные реакции или рекомбинация. В этом случае экспериментальные значения квантового выхода могут значительно превышать 1.
Последнее изменение этой страницы: 2016-08-11
lectmania.ru. Все права принадлежат авторам данных материалов. В случае нарушения авторского права напишите нам сюда...