Категории:
ДомЗдоровьеЗоологияИнформатикаИскусствоИскусствоКомпьютерыКулинарияМаркетингМатематикаМедицинаМенеджментОбразованиеПедагогикаПитомцыПрограммированиеПроизводствоПромышленностьПсихологияРазноеРелигияСоциологияСпортСтатистикаТранспортФизикаФилософияФинансыХимияХоббиЭкологияЭкономикаЭлектроника
Объединение 1-ого и 2-ого законов термодинамики. Фундаментальное уравнение Гиббса для закрытых и открытых систем.
Нер-во Клаузиуса: dS ≥ δQ/T (1). Первый закон термодинамики для открытых систем: dU= δQ+ δW + Σiμidni (*).
При изотремических процессах (1) можно записать в виде: dS= (δeQ + δiQ)/T’ (**) или dS= deS + diS, где deS – изменение энтропии, вызванное равновесным теплоообменом с окружением, diS – рост энтропии из-за необратимых процессов в системе. Подставляя (**) в (*) получим обобщенную форму записи 1 и 2 законов ТД: dU = TdS – pdV + Σiμidni или dS=1/T dU + p/T dV – 1/T Σiμidni - это фундаментальное ур-е Гиббса.
Открытая система – сществует обмен энергией и в-вом с окруж.средой.
Закрытая система – существует обмен энергией с окруж., но нет обмена в-вом.
Энтальпия: H(S,p,n) = U+pV
Функции U, H, F, G – термодинамич.потенциалы (имеют размерность энергии).
Энергия Гельмгольца: F(T,V,n) = U – TS
Энергия Гиббса: G(T,p,n) = U+pV– TS = H – TS = F + pV.
Зависимость термодин.потенциалов от их естественных переменных описывается основными ур-ями термодинамики – фунд.ур-ми Гиббса.
Фунд.ур.Гиббса для открытых систем (конспект Михаила):
dU = TdS – pdV + Σiμidni
dS=1/T dU + p/T dV – 1/T Σiμidni
dH = TdS + Vdp + Σiμidni
dF = – SdT – pdV + Σiμidni
3. Осмос. Осмотическое давление. Уравнение Вант-Гоффа и область его применимости.
Осмос– пернос растворителя ч/з полупроницаемую мембрану в сторону более концентрированных растворов.
Примеры: глаза в пресном водоеме – в глазу больше соли, вода идет в глаз, сосуды лопаются. Глаза в море – сжимаются. Питание растений.
Картинка: горизонтальный сосуд с перегородкой, пропускающей воду. В левой половине H2O, в правой H2O+NaCl. (потенциал больше слева). Вода пойдет слева направо, справа начнет повышаться давление. Слева давление p0, справа p0+π. π – избыточное – осмотрическое давление. π =сRT – формула Вант-Гоффа. (с – молярная концентрация).
Если это электролит, то π =iсRT, i – изотонический коэфф.
Приближения: раствор разбавленный, идеальный, растворитель несжимаем.
Константа скорости для реакций целого порядка и определение энергии активации по температурной зависимости константы скорости.
Реакции 0-го порядка. Скорость этих реакций не зависит от концентрации:

где [A] – концентрация исходного вещества.
Реакции 1-го порядка. В реакциях типа A → B скорость прямо пропорциональна концентрации:

Начальная концентрация [A]0 = a, текущая концентрация [A] = a – x(t), где x(t) – концентрация прореагировавшего вещества A.
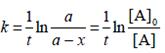
Реакции 2-го порядка. В реакциях типа A + B → D + …
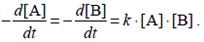
Начальные концентрации веществ: [A]0 = a, [B]0 = b; текущие концентрации: [A] = a – x(t), [B] = b – x(t). При решении этого уравнения различают два случая.
1. Одинаковые начальные концентрации веществ A и B: a = b.

2. Начальные концентрации веществ A и B различны: a ≠ b.
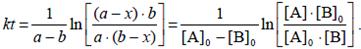
Реакции n-го порядка nA → D + …:

Ур-е Аррениуса:K(T)=A*exp[–EA/RT]
Билет 17.
Основные структурные типы ионных соединений: NaCl, CsCl, CaF2.
2. Химический потенциал компонента идеального раствора. Термодинамические функции образования идеального раствора.
Удобной системой сравнения свойств раствора является идеальный раствор – р-р, для кот.выполнено:
μi(xi)= μiø(p,T)+RTlnxi - для конденсированных р-ров
Для газов: μi(p,T)= μiø(T)+RTlnpi = μiø(T)+RTlnp + RTlnxi
μi – химический потенциал компонента в стандартном состоянии.
р – общее давление в системе.
хi – мольная доля в-ва в р-ре.
(Если неидеальный, вместо хi под ln подставлять активность). Важно: идеальные р-ры образуются из в-в с одинаковой плотностью и энергией взаимодействия м/у частицами; образуются без изменения объема.
Изменение мольных термодинамических функций при образовании бинарного идеального р-ра:
ΔfGm= RT[(1-x) ln(1-x) + xlnx],
ΔfSm= – (∂ΔfGm/∂T)p= – R{(1-x) ln(1-x) + xlnx},
ΔfHm= – T2(∂/∂T (ΔfGm/T))p=0,
ΔVm= (∂ΔfGm/∂p)T=0.
Основные понятия катализа. Классификация каталитических реакций. Гомогенный катализ. Общий механизм катализа.
Катализ – изменение скорости химической р-ии в присутсвтии катализаторов.
Катализатор – в-во, участвующее в р-ии и изменяющее ее скорость, но остающееся в первоначальном виде в конце реакции. Влияет на энергию активации, измняя путь р-ии. Не влияет на т.д.характеристики.
Основные качества: активность, селективность, устойчивость.
Классификация:
Гомогенный – реагент и катализатор в одной фазе. (Свойства: +, +, +++)
А + К ↔k-1k1 АК, АК + В →k2 Р + К.
Пример: разложение О3: Cl + O3 → ClO + O2; ClO + O3 → Cl + 2O2; 2O3 → 3O2.
1) Квазистационарное приближение. d[AK]/dt=0; k2>>k1.
r = k1k2[A][B]/(k-1+k2[B])*[K]
2) Квазистационарное приближение. k1>>k2.
r = k1k2/k -1 *[A][B][K]
Гетерогенный – –,,– в разных фазах. Реакция просходит на поверхности. (Свойства: ++, ++, +)
Пример: промышленный синтез аммиака: 3H2 + N2 →Fe 2NH3. (K равновесия уменьшается при увеличении тем-ры)
Еще пример: S + O2 →SO2, 2SO2 + O2 →V2O5 2SO3.
Ферментативный – роль катализатора играет белок. Фермент – биохимический катализатор – полимер природных аминокислот. (Свойства: +++, +++, – –)
Глобула – третичная структура белка. На ней есть ямка – активный элемент. Туда садится реагент S (substrate). E (enzyme) – фермент. P (product) – продукт. SE – фермент-субстратный комплекс.
S + E ↔k-1k1 SE →k2 Р + E.
1) k1 ~ k-1 >> k2 – квазиравновесие, первый процесс медленный (?)
2) k1 ~ k-1 << k2 – наоборот.
Уравнение Михаэлис-Ментен: скорость ферменнтативной р-ии r = rmax[S]/(KM+[S]), KM – константа Михаэлиса.
E0= [E] + [SE]
r = d[P]/dt = k2*[SE].
Для SE – метод стационарных концентраций – в большом диап.времен концентрацию этого в-ва можно считать постоянной – т.к у этого продукта маленькая концентрация.
Автокатализ – роль катализатора играет продукт р-ии.
Последнее изменение этой страницы: 2016-08-11
lectmania.ru. Все права принадлежат авторам данных материалов. В случае нарушения авторского права напишите нам сюда...