Категории:
ДомЗдоровьеЗоологияИнформатикаИскусствоИскусствоКомпьютерыКулинарияМаркетингМатематикаМедицинаМенеджментОбразованиеПедагогикаПитомцыПрограммированиеПроизводствоПромышленностьПсихологияРазноеРелигияСоциологияСпортСтатистикаТранспортФизикаФилософияФинансыХимияХоббиЭкологияЭкономикаЭлектроника
Сборка установки и проверка ее на герметичность
Установку собирают по схеме, приведенной на рисунке 1.1 (при проведении испытания в пятисекционной печи манометр перед поглотительной склянкой для аммиака не устанавливают). Всю аппаратуру соединяют встык при помощи эластичных резиновых шлангов диаметром 5 мм.
После сборки установки ее испытывают на герметичность, для чего всю систему подсоединяют к газометру и открывают винтовой зажим на шланге, отводящем воду из газометра (при закрытом манометре). При наличии герметичности вытекание воды быстро прекращается. Если же вода продолжает вытекать, необходимо найти место неплотного соединения и устранить негерметичность.
Затем газометр отсоединяют от поглотительной аппаратуры, полностью вытесняют из него воздух, заполняя насыщенным раствором хлористого натрия, после чего снова присоединяют к последней по ходу газа U-образной трубке с активированным углем. Затем отсоединяют реакционную трубку с вставкой и фильтром. Поглотительную склянку для аммиака со стороны входа газа закрывают пробкой. При этом зажимы на трубке, отводящей воду из газометра, и на манометре должны быть закрыты, а зажим на трубке, подводящей газ к газометру, должен быть открыт.
Подготовка печей при проведении испытания
В печь для коксования на слой асбеста толщиной в 1 мм перед началом опыта устанавливают пятиточечный термопреобразователь, чтобы его спаи находились в середине обогревающих секций. Печь пиролиза предварительно нагревают до (790±10) °С и выдерживают при этой температуре 30 мин, измеряя температуру термопреобразователя, установленного в середине печи.
Проведение испытания
В холодную печь коксования через печь для ватного фильтра и печь пиролиза, предварительно нагретых соответственно до (105±5) °С и (790±10) 0С, вводят реакционную трубку с кварцевой вставкой и присоединенным к ней фильтром для улавливания смолы.
Реакционную трубку в печи устанавливают так, чтобы ее конец выступал из печи пиролиза на 40 мм, после чего тотчас же соединяют фильтр с поглотительной аппаратурой, немедленно открывают зажим на шланге, отводящем воду из газометра, а затем на манометре. В течение всего опыта поддерживают разрежение в газометре 98—196 Па.
Температуру в печи пиролиза и печи для ватного фильтра доводят до первоначальной и включают командное устройство, подающее напряжение на секции печи коксования, при этом первой включается секция, прилегающая к печи пиролиза. Через 15 мин автоматически включается вторая секция; затем последовательно через каждые 10 мин включается третья, четвертая, пятая секции печи коксования. После включения пятой секции командное устройство автоматически выключается, оставив цепи управления всех секций коксования включенными.
После нагрева последней пятой секции печи коксования до (890±10) 0С во всех секциях выдерживают температуру (890±10) °С, а в печи пиролиза (790±10) °С в течение 20 мин, после чего опыт заканчивают и выключают обогрев печей.
Температура секций печи коксования (890±10) 0С и печи пиролиза (790±10) 0С поддерживается автоматически при помощи регулирующих милливольтметров и контактного реле с ртутными переключателями.
Втечение опыта каждые 5 мин записывают температуру секций печей коксования и пиролиза, количество и температуру газа в газометре.
К концу опыта в газометре уравнивают давление с атмосферным, что достигается регулированием оттока воды из газометра, после чего закрывают зажимы на шланге, отводящем воду из газометра, манометре и шланге, соединяющем поглотительную систему с газометром, определяют объем выделившегося газа по объему раствора, вытекшего из газометра, и записывают барометрическое давление.
После окончания опыта отсоединяют фильтр для улавливания смолы от остальной поглотительной аппаратуры и вынимают термометр из печи с ватным фильтром. Реакционную трубку вместе со вставкой и фильтром вынимают из печей и, положив на лист толстого асбеста, отсоединяют кварцевую вставку и фильтр. Реакционную трубку плотно закрывают резиновой пробкой во избежание выгорания кокса и охлаждают до комнатной температуры, затем выдерживают в течение 20 мин в открытом состоянии и взвешивают на технических весах.
Кварцевую вставку и фильтр охлаждают до комнатной температуры в эксикаторе. Всю поглотительную аппаратуру разъединяют и оставляют открытой на 20 мин на воздухе, кроме U-образных трубок с хлористым кальцием и активированным углем, которые выдерживают в открытом состоянии в эксикаторе. Затем кварцевую вставку, фильтр, поглотительные склянки, U-образные трубки с хлористым кальцием и трубки с активированным углем взвешивают на аналитических весах с точностью до 0,0002 г.
Из кварцевой трубки извлекают насадку и коксовый остаток. Трубку и насадку помещают в трубчатую печь, нагретую до 700—800 °С, для выжигания графита и мелких кусочков кокса; вставку и фильтр, освобожденный от ваты, прокаливают в той же печи.
Обработка результатов
1) Выход кокса определяют по массе коксового остатка в кварцевой трубке. Выход кокса (X) в процентах вычисляют по формуле
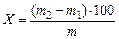
где m2 — масса кварцевой трубки с коксовым остатком после опыта, г;
m1 — масса кварцевой трубки до загрузки пробы, г;
m — масса навески угля, г.
За результат анализа принимают среднее арифметическое двух параллельных определений, расхождение между которыми при доверительной вероятности Р = 0,95 не должно превышать 0,5 % абс.
2) Выход смолы определяют по изменению массы вставки, фильтра и массе легких смоляных погонов, извлеченных эфиром из поглотительного раствора для аммиака.
Выход легких смоляных погонов определяют следующим образом: поглотительный раствор для аммиака из поглотительной склянки переводят в делительную воронку вместимостью 25 см3, а поглотительную склянку тщательно промывают небольшими порциями (по 1,5 — 2 см3) петролейного эфира. Промывание производят 2 — 3 раза до полного растворения смоляных продуктов, переводят эфир в ту же делительную воронку и перемешивают взбалтыванием с поглотительным раствором. Поглотительный раствор из делительной воронки осторожно декантируют в коническую колбу вместимостью 250 см3 и сохраняют для дальнейшего определения аммиака. Поглотительную склянку промывают до нейтральной реакции небольшими порциями дистиллированной воды, промывая этой же водой и эфирные вытяжки в делительной воронке.
Промывные воды переводят в ту же колбу с поглотительным раствором. Эфирные вытяжки переводят в предварительно взвешенную коническую колбу вместимостью 25 см3. Петролейный эфир отгоняют из колбы под вакуумом (водоструйный насос) при охлаждении колбы льдом, пропуская через нее осушенный воздух. Для этой цели служит прибор, изображенный на рисунке 1.3. После отгонки эфира колбу тщательно вытирают снаружи и взвешивают.

1 — трубка с хлористым кальцием; 2 — колба с эфирной вытяжкой; 3 — водяная баня; 4 — колба Бунзена.
Рисунок 1.3
Выход смолы (X1) в процентах вычисляют по формуле

где т1 — масса смолы в фильтре и вставке, г;
т2— масса смолы, извлеченная эфиром, г;
т — масса навески угля, г.
За результат анализа принимают среднее арифметическое двух параллельных определений, расхождение между которыми при доверительной вероятности Р = 0,95 не должно превышать, указанных в таблице 1.1.
Таблица 1.1
| Массовая доля смолы, % | Допускаемое расхождение, % абс. |
| От 0,10 до 0,3 Св. 0,3 » 1,0 » 1,0 » 3,0 » 3,0 » 10 | 0,1 0,2 0,3 0,4 |
3) Выход аммиака определяют по объему раствора едкого натра концентрации эквивалента 1 моль/дм3, израсходованного на титрование поглотительного раствора после извлечения из него легких смоляных погонов.
Выход аммиака (Х2) в процентах вычисляют по формуле
 ,
,
где V— объем раствора серной кислоты концентрации эквивалента 0,1 моль/дм3, взятый для опыта, см3;
V1 — объем раствора щелочи концентрации эквивалента 0,1 моль/дм3, израсходованный на титрование избытка серной кислоты, не вступившей в реакцию, см3;
Т — титр раствора серной кислоты концентрации эквивалента 0,1 моль/дм3, выраженный по аммиаку, г/см3;
m — масса навески угля, г.
За результат анализа принимают среднее арифметическое двух параллельных определений, расхождение между которыми при доверительной вероятности Р = 0,95 не должно превышать значений, указанных в таблице 1.2.
Таблица 1.2
| Массовая доля аммиака, % | Допускаемое расхождение, % абс. |
| От 0,005 до 0,015 Св. 0,015 » 0,045 » 0,045 » 0,130 » 0,13 » 0,40 » 0,40 » 1,0 | 0,004 0,010 0,020 0,03 0,05 |
4) Выход общей влаги определяют по разности между увеличением массы поглотителя для аммиака и трубки с хлористым кальцием, помещаемой за ним во время опыта, и массой аммиака и смоляных погонов, извлеченных эфиром.
Выход общей влаги (Х3) в процентах вычисляют по формуле
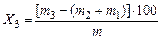 ,
,
где m3— увеличение массы поглотителя для аммиака и трубки с хлористым кальцием, г;
m2 — масса смолы, извлеченная эфиром, г;
m1 — масса аммиака, г;
т — масса навески угля, г.
За результат анализа принимают среднее арифметическое двух параллельных определений, расхождение между которыми при доверительной вероятности Р = 0,95 не должно превышать 0,2 % абс., при массовой доле общей влаги от 1 до 3 %, и 0,3 % абс., при массовой доле обшей влаги от 3 до 10 %.
5) Выход пирогенетической воды определяют по разности между выходом общей влаги и содержанием влаги в анализируемой пробе угля.
Выход пирогенетической воды (Х4) в процентах вычисляют по формуле
 ,
,
где Х4— выход общей влаги, %;
Wa— содержание влаги в аналитической пробе угля, %.
6) Выход сероводорода определяют следующим образом. Щелочной раствор из поглотительной склянки переводят в мерную колбу вместимостью 250 см3, а поглотительную склянку промывают два раза дистиллированной водой. Промывные воды сливают в ту же мерную колбу, доводят уровень раствора дистиллированной водой до метки и перемешивают. Затем в коническую колбу вместимостью 250 см3 наливают 50 см3 дистиллированной воды, 25 см3 испытуемого раствора и вводят для подкисления раствора разбавленную 1:1 соляную кислоту, количество которой устанавливают заранее (в отдельную порцию — 25 см3 испытуемого раствора добавляют 50 см3 дистиллированной воды, вводят метиловый оранжевый, титруют до нейтральной реакции, разбавленной 1:1 соляной кислотой и дают в избыток 2 — 4 капли той же кислоты). Из бюретки приливают 10 — 30 см3 раствора йода концентрации эквивалента 0,1 моль/дм3 (в зависимости от содержания серы в угле). Избыток йода, не вступивший в реакцию с сероводородом, титруют 0,1 н. раствором тиосульфата натрия концентрации эквивалента 0,1 моль/дм3 в присутствии крахмала, который вводят под конец титрования.
Выход сероводорода (Х5) в процентах вычисляют по формуле
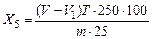 ,
,
где V - объем раствора йода концентрации эквивалента 0,1 моль/дм3, взятый для анализа, см3;
V1 - объем раствора тиосульфата натрия концентрации эквивалента 0,1 моль/дм3, израсходованный на титрование избытка йода, не вступившего в реакцию с сероводородом, см3;
T - титр раствора йода концентрации эквивалента 0,1 моль/дм3, выраженный по сероводороду,г/см3;
250 - общий объем испытуемого раствора, см3;
m — масса навески угля, г;
25 — аликвотная часть раствора, см3.
7) Выход диоксида углерода определяют по разности между увеличением массы поглотителя для сероводорода и диоксида углерода и трубки с хлористым кальцием, помещаемой за ним во время опыта, и количеством определенного сероводорода.
Выход диоксида углерода (Х6) в процентах вычисляют по формуле
 ,
,
где m1, — увеличение массы поглотительной склянки с раствором щелочи и трубки с хлористым кальцием, г;
m — масса навески угля, г;
Х5— выход сероводорода, %.
8) Выход непредельных углеводородов определяют по увеличению массы поглотительной склянки с раствором желтого оксида ртути в серной кислоте и трубки с хлористым кальцием, помещаемой за ней во время опыта.
Выход непредельных углеводородов (Х7) в процентах вычисляют по формуле
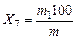 ,
,
где т1 — увеличение массы поглотительной склянки с раствором для поглощения непредельных углеводородов и трубки с хлористым кальцием, г;
т — масса навески угля, г.
9) Выход сырого бензола определяют по увеличению массы трех U-образных трубок с активированным углем.
Выход сырого бензола (X8) в процентах вычисляют по формуле
 ,
,
где т1— увеличение массы трех трубок с активированным углем, г;
т — масса навески угля, г.
За результат анализа принимают среднее арифметическое двух параллельных определений, расхождение между которыми при доверительной вероятности Р = 0,95 не должно превышать 0,05 % абс., при выходе сырого бензола от ,1 до 0,3 %, и 0,1 % абс., при выходе сырого бензола от 0,3 до 1,0 %.
10) Выход выделившегося в условиях опыта газа определяют по объему раствора, вытекшего из газометра.
Выход выделившегося газа, приведенного к нормальным условиям, (Х9), в м3/т вычисляют по формуле
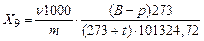 ,
,
где ν — объем выделившегося газа, определенный по объему раствора, вытекшего из газометра, дм3;
В — барометрическое давление во время проведения опыта, Па;
р — упругость водяных паров при данной температуре газа, Па;
m — масса навески угля, г;
t — температура газа в газометре, 0С.
За результат анализа принимают среднее арифметическое двух параллельных определений, расхождение между которыми при доверительной вероятности Р = 0,95 не должно превышать 5 м3/т.
11) Для вычисления общего выхода газа предварительно производят расчет выхода сероводорода (X10), диоксида углерода (Х11) и непредельных углеводородов (Х12) в м3/т по следующим формулам:
 ,
,  ,
,  ,
,
где Х5, Х6, X7 — выход соответственно сероводорода, диоксида углерода и непредельных углеводородов, %;
ρ1, ρ2, ρ3 — масса 1 м3 соответственно сероводорода, диоксида углерода и непредельных углеводородов, кг/м3.
12) Общий выход газа при нормальных условиях (Х13) в м3/т вычисляют по формуле
 ,
,
где Х9— выход выделившегося газа без учета выхода сероводорода, диоксида углерода и непредельных углеводородов, м /т;
X10, Х11, Х12 — выход соответственно сероводорода, диоксида углерода и непредельных углеводородов, м3/т.
13) Пересчет выхода продуктов коксования на сухую массу (Вс) в процентах производят по формуле
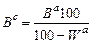 ,
,
где Вa — выход данного продукта на аналитическую пробу, %;
Wa — содержание влаги в аналитической пробе, %.
14) Вычисление результатов определения выхода продуктов коксования производят до третьего десятичного знака и округляют до второго, а выход газа — до первого десятичного знака и округляют до целого числа.
Вопросы
1. Укажите области применения сырого бензола, смолы и коксового газа. Каковы требования к их качеству?
2. Сравните состав смолы коксования и полукоксования.
3. Как влияет увеличение в шихте количества газовых углей на выход и качество летучих продуктов коксования?
4. Почему необходимо улавливать аммиак и сероводород из коксового газа?
5. Какое влияние оказывает уровень обогрева коксовых печей на состав продуктов обогрева?
6. Как изменится состав химических продуктов при коксовании термически подготовленной шихты?
МЕТОД ОПРЕДЕЛЕНИЯ ОБЪЕМНОЙ ДОЛИ КОМПОНЕНТОВ НА КОМПЛЕКТАХ ДЛЯ ГАЗОВЫХ АНАЛИЗОВ ТИПА КГА
Общие сведения
Настоящий стандарт устанавливает метод определения объемной доли компонентов в газах на комплектах для газовых анализов типов КГА2 и КГАЗ.
Сущность метода заключается в определении:
1) суммы кислотообразующих газов (СО2, SО2, H2S и др.);
2) суммы непредельных углеводородов (СnН2n), О2 и СО - абсорбционным избирательным поглощением поглотительными растворами;
3) Н2 - сжиганием с палладиевым катализатором;
4) суммы предельных углеводородов (СnН2n+2) и Н2 - фракционным сжиганием над окисью меди, при этом возможно раздельное определение СН4 и суммы его гомологов, принимаемых за С2Н6.
Средства измерения
При определении объемной доли компонентов в газовых смесях применяют следующую аппаратуру и реактивы:
1) комплекты для газовых анализов типов КГА2 и КГАЗ;
2) термопару хромель-алюмель и милливольтметр с градуировкой шкалы до 1000 °С.
Подготовка прибора к анализу
Схема установки приведена на рисунке 1.
Стеклянный цилиндр 1заполняют дистиллированной водой через отверстие в пробке. При этом во избежание образования пузырьков воздуха на стенках бюретки вода через резиновую трубку должна поступать на дно цилиндра. По заполнении цилиндра отверстие закрывают пробкой или стеклянной заглушкой.
Бюретку 3заполняют дистиллированной водой. Для этого воду наливают в уравнительную склянку 30и, перекрыв кран 27на сообщение бюретки с атмосферой через правую трубку гребенки (заглушку крана 28снять) при открытых кранах вилки 4,поднимают уравнительную склянку 30и вытесняют из бюретки 3воздух, заполняя ее водой до метки «0». Вслед за этим краны вилки 4закрывают, кран бюретки 27переключают на сообщение с левой частью гребенки.
После этого приступают к заполнению сосудов поглотительными растворами.
Все сосуды, кроме сосуда 11, заполняют, не снимая с прибора, через воронку, вставленную в отверстие пробки. Заполнив вспомогательную камеру, кран сосуда перекрывают на сообщение с бюреткой и, опустив уравнительную склянку, при открытых кранах вилки переводят раствор в поглотительную камеру до метки. Затем все краны и уравнительную склянку ставят в первоначальное положение. При этом слой раствора во вспомогательной камере должен быть высотой 30-40 мм. Сосуд 10заполняют раствором гидроокиси калия.

1 – стеклянный цилиндр; 2 – компенсационная трубка; 3 - бюретка; 4 - вилка; 5 – компенсатор давления; 6 – трубка с катализатором; 7 – источник электрического тока; 8 - кран; 9 – поглотительный сосуд без наполнителя; 10 - 13 – поглотительные сосуды с насадкой; 14, 16 – 19, 27, 28 – серповидный кран; 15 - гребенка; 20 – трубка для сжигания; 21 - электропечь; 22 - термометр; 23 – источник электрического тока; 24 – прямые краны; 25 – трехходовой кран; 26 - кран; 29 - заглушки; 30 – уравнительная склянка.
Рисунок 1
Сосуд 11снимают с прибора и наполняют его раствором брома в вытяжном шкафу. Отверстие сосуда закрывают предохранительной трубкой с шарообразным расширением, заполненным аскаритом и закрытым с обеих сторон пробочками из ваты. По заполнении сосуд устанавливают в прибор и переводят раствор в поглотительную камеру.
Сосуд 12 заполняют раствором пирогаллола.
Сосуд 13заполняют суспензией закиси меди в серной кислоте. После заполнения сосудов 12и 13в отверстие пробок вставляют отросток гребенки 15, на свободный конец которой надевают резиновый баллон-мешочек из кислотощелочестойкой резины или присоединяют заполненную водой склянку.
Сосуд 9без насадки заполняют насыщенным раствором хлористого натрия или другим раствором, указанным в п. 1.1.
Трубку для сжигания 20в той части, которая погружается в печь 21, заполняют гранулированной окисью меди. Загрузку ведут в оба конца трубки, слегка утрамбовывая постукиванием. Глубину загрузки контролируют введением гибкой стальной проволоки. Чтобы окись меди не высыпалась при перевертывании трубки, в оба конца закладывают неплотные асбестовые пробочки или спиральки из медной проволоки.
Трубку с катализатором 6между контактами заполняют палладиевым катализатором и с двух концов закрывают асбестовыми пробками или стекловатой.
После этого трубки для сжигания присоединяют к прибору при помощи толстостенных резиновых трубок.
Герметичность прибора проверяют следующим способом.
Перекрыв кран 27бюретки 3на сообщение с воздухом (через верхнюю часть гребенки), поднимают воду в ней до крана. Затем перекрывают кран 27на сообщение бюретки с левой частью гребенки, на конец которой надета заглушка. Открыв один из кранов вилки 4, опускают уравнительную склянку 30на всю длину резиновой трубки. Если уровень жидкости в бюретке, несколько снизившись при опускании склянки, останется затем неподвижным и уровни в поглотительных сосудах также останутся неподвижными в течение 5-8 мин, прибор герметичен. Если же существуют какие-либо неплотности, уровень в бюретке будет постепенно понижаться. Для установления причины неплотности прибор испытывают по частям. Сначала перекрывают гребенку краном 25. Если прибор в таком положении сохраняет герметичность, то кран 25 ставят в прежнее положение, гребенку же перекрывают краном 19, повернув его на 60° в ту или другую сторону и вновь испытывают на герметичность. Перекрывая постепенно один за другим все остальные краны на 60°, находят участок, создающий неплотность прибора, и устраняют причину неплотности. Проверку на герметичность проводят также с включенной в систему трубкой для сжигания 20и трубкой с катализатором 6 с приемником газа 9.
Если указанным способом причина неплотности не выяснена, то гребенку заполняют водой и, проверяя на герметичность, наблюдают появление пузырьков воздуха в месте неплотности.
После замены непригодной детали воду удаляют, гребенку промывают этиловым спиртом или этиловым эфиром через воронку, присоединяя резиновую трубку к одному из концов гребенки, и просушивают струей воздуха.
После проверки прибора на герметичность дистиллированную воду в бюретке заменяют запирающей жидкостью.
Влияние вредного пространства гребенки и трубок для сжигания на измеряемый объем анализируемого газа исключают следующим образом.
В бюретку 3берут азот, очищенный от кислорода, и, сняв заглушку с левого отростка гребенки (кран 14),продувают азотом сначала гребенку, затем трубки для сжигания 20 и 6 при открытом кране 8, перекрывая соответственно краны. Подняв запирающую жидкость в бюретке 3до метки «0», закрывают краны вилки 4. Затем, перекрыв кран 25манометра на сообщение с прибором, устанавливают одинаковое давление во всех частях прибора. Когда жидкость в манометре будет на одном уровне, закрывают сначала краны вилки 4, затем кран 25манометра, а также одноходовые краны 24на отростках трубки для сжигания 20.
Вредное пространство гребенки и трубок для сжигания, заполненное азотом, перед и после анализа должно быть исключено из общего объема анализируемого газа. Объем отростка бюретки от «0» до крана 27устанавливают экспериментально или расчетом; этот объем добавляют к замеренному объему анализируемого газа.
Незначительным объемом крановых отростков над поглотительными растворами пренебрегают.
Нагрев электрической печи 21предварительно устанавливают с помощью автотрансформатора на температуру 260-270 °С - для сжигания водорода и 850-950°С - для сжигания предельных углеводородов.
На разогрев печи до указанных температур должно затрачиваться 7-10 мин. Температура печи должна поддерживаться в указанных пределах в течение 10-15 мин. Температура 260-270 °С как при регулировке, так и в процессе анализа, контролируется термометром 22.
Нагрев до 850-950 °С (светло-красное каление) контролируется термопарой только при регулировке.
Проведение испытания
Последнее изменение этой страницы: 2016-07-22
lectmania.ru. Все права принадлежат авторам данных материалов. В случае нарушения авторского права напишите нам сюда...